Chemcraft破解版是功能强大的是用于量子化学计算程序,软件支持通过原子坐标绘制分子的3维图像,并可以检查或修改分子中的任何几何参数(距离,角度)。提供构建和修改分子几何形状的工具,您可以使用标准分子片段,并拖动图像中的原子和片段进行相关操作,非
Chemcraft破解版是功能强大的是用于量子化学计算程序,软件支持通过原子坐标绘制分子的3维图像,并可以检查或修改分子中的任何几何参数(距离,角度)。提供构建和修改分子几何形状的工具,您可以使用标准分子片段,并拖动图像中的原子和片段进行相关操作,非常方便,这款强大的图形化程序使用将为用户带来方便的可视化计算操作,并快速得到准确的结果,为您的工作带来更大的便捷,软件支持处理各种计算类型,支持使用以文本格式导入/导出原子坐标。虽然说Chemcraft本身不执行计算,但是可以大大方便使用广泛的量子化学软件包。本次带来最新破解版下载,含注册码,有需要的朋友不要错过了!
功能特色
1、通过原子坐标绘制分子的3维图像,并可以检查或修改分子中的任何几何参数(距离,角度);
2、游戏,高斯,NWChem,ADF,Molpro,Dalton,Jaguar,Orca,QChem输出文件的可视化:文件中各个几何的表示(优化结构,每个优化步骤的几何等),振动模式动画,图形表示梯度(作用在核上),等值面或有色平面形式的分子轨道的可视化,振动或电子(例如TDDFT)光谱的可视化,显示SCF收敛图的可能性以及其他一些特征;
3、用于构建分子和修改分子几何形状的不同工具:使用标准分子片段,“拖动”分子图像中的原子或片段,设置对称点组的实用程序以及其他可能性;
4、以可定制的显示模式制作分子的出版物就绪图像,其中包含所需的名称(标签,线条等);
5、用于准备输入文件的一些附加实用程序:构造Z矩阵,自动生成具有非标准基集的输入文件,将从输出文件读取的MO转换为输入文件的格式。
6、该程序结合了先进的图形用户界面和为实际使用而设计的有益功能。Chemcraft基于将文件划分为单独的元素并将其显示在分层的多级列表中,从而提供了非常详细的输出文件的结构化可视化;此功能使您可以轻松分析复杂的文件,例如扫描作业,IRC作业或多作业计算。Chemcraft的图形引擎不需要任何硬件加速。
7、Chemcraft是一种商业软件。Windows版本的价格目前对于学术用户为160美元,对于其他用户为300美元。也可以下载Chemcraft for Windows的免费软件版本,它具有一些限制和导航屏幕。
使用说明
1、Chemcraft基础
Chemcraft包含一组图形工具,可帮助进行量子化学计算。它提供了方便的实用程序,可帮助准备新作业以进行计算和分析计算结果。该程序的主要功能是可视化量子化学软件包产生的输出文件。支持的主要软件包是Games(美国版和PCGames)和Gaussian94-09。
2、Chemcraft游戏用户
Chemcraft提供了Games-US输出文件的非常详细的可视化。文件中的以下数据可以图形方式显示:
-原子坐标(如果文件中存在相应的表,则对应于全部或对称唯一原子);
-如果在文件中提供了键顺序分析,则文件中的键会显示在图像上(否则,键是通过距离算法计算的);
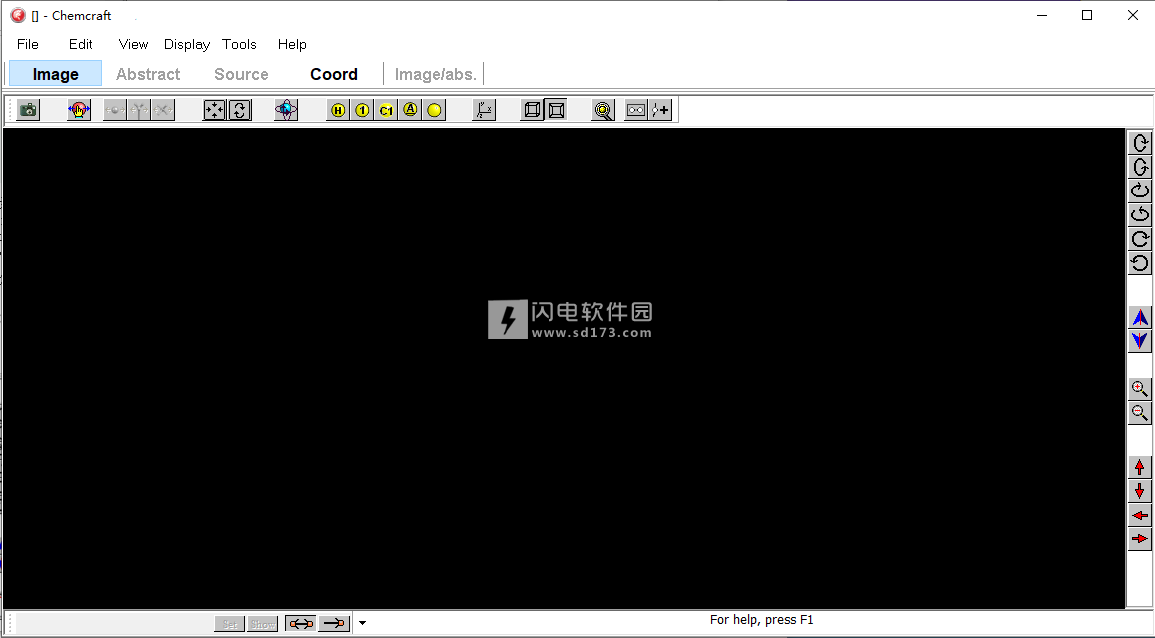
-能量梯度可以以指针的形式显示(图2);
-不同的原子特性可以显示为原子上的标记:穆里肯数和电荷,自旋密度,化合价;
-债券定单(作为债券上的标签);
-分子轨道可视化为等值面或彩色平面;
-振动模式可以设置动画或以指针(位移矢量)的形式显示;
-偶极矩可以可视化为指针;
-MO能量可以以图的形式显示。
该程序提供输出文件的结构化表示。读取的文件分为单独的元素,例如单独的几何形状或振动模式。对于每个元素,将从文件中提取所有可用数据:原子坐标,能量梯度等。所有元素都以层次结构列表的形式显示(见图1)。单击列表的元素会自动在图像上显示各个几何形状或模式,并允许可视化不同的属性。该界面可提供可靠的计算数据可视化,包括非标准类型的计算,不完整的计算等。它还可以可视化具有多个计算作业的复杂文件。对于能量扫描或IRC计算,所有几何形状均按扫描/IRC步骤分为几组。
能量梯度可视化示例。
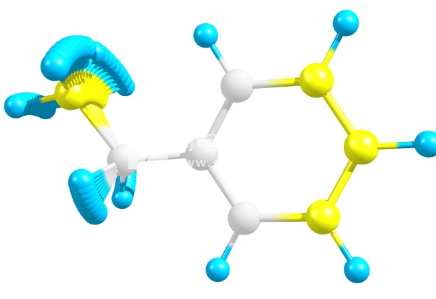
Chemcraft从GAMESS-US输出文件中提取分子轨道系数以及基本信息,并以等密度面或以密度值着色的表面(平面,球体)的形式绘制分子轨道(见图3)。Chemcraft提供了一些执行轨道操作的可能性(例如,将一个轨道与另一个轨道相乘)。建立轨道的公式取自随GAMESS-US发行的PLTORB程序的源代码。具有密度值的多维数据集的计算得到了很好的优化。请注意,如果文件中有多个分子轨道系数表,则Chemcraft会提取它们中的每一个并允许对其进行渲染(例如,在MCSCF计算中,可以显示标准轨道或自然轨道)。除了可视化轨道,

分子轨道可视化的示例。
Chemcraft支持一个界面,用于快速创建带有非标准基础集的GAMESS-US输入文件的各个部分(图4)。基集是从它们的描述中提取的,可以从PNNL的网页(http://www.emsl.pnl.gov/forms/basisform.html)中获得。它们还可以由用户指定的其他高斯来补充。
3、适用于高斯用户的Chemcraft
我们建议在高斯输入文件中键入#P GFINPUT POP(FULL,NBO),以便通过Chemcraft可视化高斯输出。#P选项启用扩展的打印输出;GFINPUT选项启用基集信息的打印输出(基集中基元的描述),而POP(FULL)启用所有分子轨道系数的打印输出(也可以使用POP(REGULAR))。后两个关键字允许Chemcraft可视化分子轨道。POP(NBO)可以打印出“自然键轨道”分析,其中可以计算分子中的键。所有这些关键字都是可取的,但不是必需的。对于GAMESS文件,可以看到文件中的不同数据:作用在核上的力(能量梯度),原子电荷,自旋密度和其他原子特性,NBO键的特性(占有率,能量),正常模式,分子轨道(可以显示笛卡尔(6d等)或内部(5d)函数),MO能量。从文件中读取标准或输入/ Z矩阵方向的坐标,并将其显示在图像上(这对于正确可视化作用在核上的力是必需的,因为它们通常以与其他属性不同的方向打印)。对于能量表面扫描和IRC作业,所有几何形状均按扫描步骤分组。对于每个单独的几何形状或振动模式,概述了最基本的数据并将其显示为“抽象”(SCF能量,收敛准则等)。Chemcraft读取多步高斯作业,然后将其显示为几个扩展节点的列表,每个节点代表文件中的单个作业。等)或内部(5d)功能可以可视化),MO能量。从文件中读取标准或输入/ Z矩阵方向的坐标,并将其显示在图像上(这对于正确可视化作用在核上的力是必需的,因为它们通常以与其他属性不同的方向打印)。对于能量表面扫描和IRC作业,所有几何形状均按扫描步骤分组。对于每个单独的几何形状或振动模式,概述了最基本的数据并将其显示为“抽象”(SCF能量,收敛准则等)。Chemcraft读取多步高斯作业,然后将其显示为几个扩展节点的列表,每个节点代表文件中的单个作业。等)或内部(5d)功能可以可视化),MO能量。从文件中读取标准或输入/ Z矩阵方向的坐标,并将其显示在图像上(这对于正确可视化作用在核上的力是必需的,因为它们通常以与其他属性不同的方向打印)。对于能量表面扫描和IRC作业,所有几何形状均按扫描步骤分组。对于每个单独的几何形状或振动模式,概述了最基本的数据并将其显示为“抽象”(SCF能量,收敛准则等)。Chemcraft读取多步高斯作业,然后将其显示为几个扩展节点的列表,每个节点代表文件中的单个作业。从文件中读取标准或输入/ Z矩阵方向的坐标,并将其显示在图像上(这对于正确可视化作用在核上的力是必需的,因为它们通常以与其他属性不同的方向打印)。对于能量表面扫描和IRC作业,所有几何形状均按扫描步骤分组。对于每个单独的几何形状或振动模式,概述了最基本的数据并将其显示为“抽象”(SCF能量,收敛准则等)。Chemcraft读取多步高斯作业,然后将其显示为几个扩展节点的列表,每个节点代表文件中的单个作业。从文件中读取标准或输入/ Z矩阵方向的坐标,并将其显示在图像上(这对于正确可视化作用在核上的力是必需的,因为它们通常以与其他属性不同的方向打印)。对于能量表面扫描和IRC作业,所有几何形状均按扫描步骤分组。对于每个单独的几何形状或振动模式,概述了最基本的数据并将其显示为“抽象”(SCF能量,收敛准则等)。Chemcraft读取多步高斯作业,然后将其显示为几个扩展节点的列表,每个节点代表文件中的单个作业。对于能量表面扫描和IRC作业,所有几何形状均按扫描步骤分组。对于每个单独的几何形状或振动模式,概述了最基本的数据并将其显示为“抽象”(SCF能量,收敛准则等)。Chemcraft读取多步高斯作业,然后将其显示为几个扩展节点的列表,每个节点代表文件中的单个作业。对于能量表面扫描和IRC作业,所有几何形状均按扫描步骤分组。对于每个单独的几何形状或振动模式,概述了最基本的数据并将其显示为“抽象”(SCF能量,收敛准则等)。Chemcraft读取多步高斯作业,然后将其显示为几个扩展节点的列表,每个节点代表文件中的单个作业。
除了高斯输出文件外,Chemcraft还可以读取格式化的检查点文件(.fch),从文件中提取分子结构和轨道。为了可视化分子轨道和其他特性,还可以读取高斯立方体文件。
Chemcraft使用NMR计算(GIAO,CSGT)从高斯日志文件中读取各向同性屏蔽值。提供了一种简单的实用程序,用于将其重新计算为化学位移并在指定的原子组内求平均值。
4、适用于ADF用户的Chemcraft
Chemcraft读取ADF输出文件,ASCII TAPE21文件和ASCII TAPE41文件。目前,ADF输出文件的可视化不如GAMESS或高斯文件的可视化:提供能量提取,偶极矩和某些原子特性的可视化,但没有分子轨道的可视化等。
5、使用其他格式
除了Games-US,Gaussian和ADF文件之外,Chemcraft还可以读取NWChem,Jaguar,Orca,Dalton,GAMESS-UK,Turbomole,Molpro和QChem输出文件,HyperChem文件,MSI或PDB格式的文件(不全面支持这些格式)以及MolDraw和Priroda程序格式,NBO格式(.31-.40文件),Molden文件,MFJ,SDF和Tinker文件,Crystal,VASP和Shellx文件,晶体学CIF文件以及具有原子笛卡尔坐标的简单文本文件。Chemcraft提供了一个界面,可通过剪贴板以文本格式导入/导出原子坐标,这有助于使用任何类型的计算中的数据。将原子坐标导出到剪贴板对于快速创建输入文件也很有用。
Chemcraft包含一个实用程序,可用于将晶体学测量中使用的分数坐标转换为笛卡尔坐标,以及反之,使用晶胞参数(a,b,c等)。
6、构造分子
Chemcraft支持一套用于构建分子结构的工具,可用于准备用于计算或其他目的的初步猜测:
-从标准分子片段(自由基等)构建分子。提供了用自定义片段补充片段集并通过剪贴板复制/粘贴单个片段的可能性;
-修改分子中的任何几何参数(距离,角度,二面角)。所述修饰可以伴随一个原子,两个原子或所选原子组的置换;
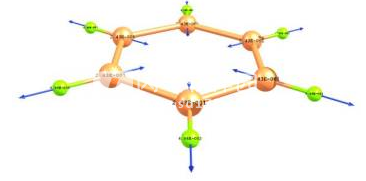
-使用鼠标“拖动”原子或片段在分子图像上或旋转片段的可能性(图5);
-用于应用任意几何参数集的迭代算法(图6);
-易于使用的工具,可将点基应用于分子(图8)。
拖动”原子或沿键旋转片段的示例。
当“拖动”原子或执行其他结构修改时,可以在图像上控制任何几何参数(参见图5)。Chemcraft的界面允许人们轻松更改任何原子的类型或插入/除去键。
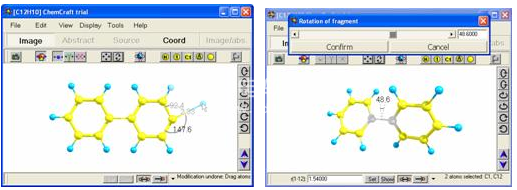
Chemcraft提供了一种用于快速获取z矩阵的实用程序。通过单击分子中的原子并指定一些其他信息来构建z矩阵(图7)。在使用该实用程序之前,应首先获取笛卡尔坐标中的分子结构。所有上述用于构建分子的工具均可用于此目的。
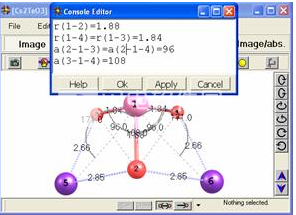
应用一组指定的几何参数。
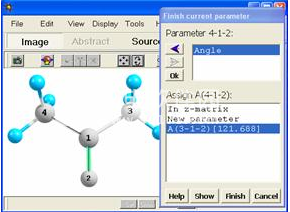
Z矩阵的可视化构造。
Chemcraft可以调整分子中的原子坐标以应用指定的对称性(图8)。此实用程序在您的研究中非常有用,因为对称分子的计算速度比非对称分子快。
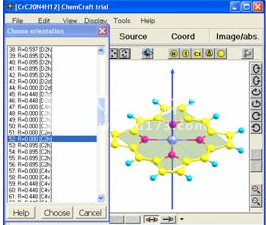
应用对称点组
7、合并分子结构
Chemcraft支持一套用于构建分子结构的工具,可用于准备用于计算或其他目的的初步猜测:
合并多个结构
在使用此实用程序之前,应选择几个原子以使其彼此最大距离放置(在图9中,这些原子标记为白色)。
除此功能外,Chemcraft还可以计算两个分子之间的均方根差。
8、分子渲染的可能性
Chemcraft可以生成分子的高质量32位图片。它被设计为用于创建发布就绪图像的程序,不需要任何其他修改。可以在原子/键和其他对象(例如标签和线条)上轻松添加图片标题来补充图片。Chemcraft包含一组标准显示方案。每个方案代表一组定义分子外观的参数:照明参数,单个原子和键的颜色和大小等。图10展示了该集合的四个方案。Chemcraft允许用户更改单个方案的参数或将自己的方案添加到集合中。
Chemcraft的图形引擎不需要任何图形加速或其他图形库。它经过了优化,即使在过时的计算机上也可以提供很高的渲染速度。
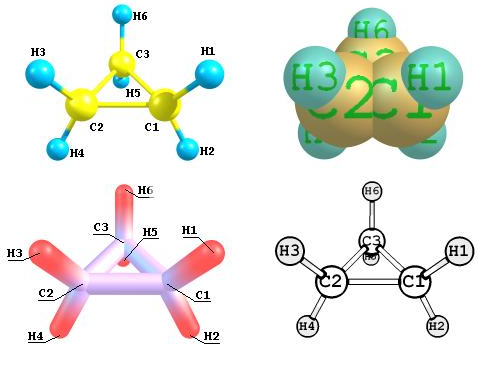
用不同的显示方案和不同的标签样式渲染分子的示例。
9、创建动画
Windows版本的Chemcraft可以以多个位图文件或gif动画文件的形式保存分子的动画图片。提供以下标准动画类型:分子旋转动画,振动频率和PES扫描动画。以下动画是使用Chemcraft创建的:
2025最新版联系 1400155802@qq.com
 Chemcraft 1.8 Build 760b 2025 破解版
Chemcraft 1.8 Build 760b 2025 破解版
 牙科CAD设计软件 exocad Dental
牙科CAD设计软件 exocad Dental Synopsys CoreTools vW-2024.09-
Synopsys CoreTools vW-2024.09- AviCAD 2025 Pro 25.0.10.5 x64
AviCAD 2025 Pro 25.0.10.5 x64  Synchro plus SimTraffic 12.2.5
Synchro plus SimTraffic 12.2.5 Kenny Asset Forge 2.5.0
Kenny Asset Forge 2.5.0 CPFD Barracuda Virtual Reactor
CPFD Barracuda Virtual Reactor